
In Situ-generated Vaccine-like Pyroptosome for Personalized Cancer Immunotherapy: Insights from CT26-OVA Cells
1. Research Background
Cancer vaccines are frontier strategies in immunotherapy, with potential in activating adaptive immune responses and enhancing anti-tumor immunity. Despite progress in preclinical and clinical research over decades, they have not become a landmark tumor therapy. Vaccine efficacy depends on immunogenicity, which is determined by the combination of antigen and adjuvant. Tumor antigen heterogeneity between tumors and patients necessitates the development of personalized neoantigen vaccines. Genomic sequencing, machine learning and mRNA technology have brought encouraging results for personalized vaccines, but high cost and complex production limit their wide application.
In situ cancer vaccines need to synergistically activate the CIC, which includes antigen release from dying cancer cells, antigen processing and presentation by APCs, and generation of tumor-specific effector T cells to lyse tumor cells. However, the rate-limiting capacity of each step in CIC and the immunosuppressive tumor microenvironment hinder effective CIC activation and subsequent anti-tumor immune responses. Multiple strategies have been developed to amplify CIC, including hybridization with bacterial membranes, synergy with fluoropolymers, combination with immune adjuvants, integration with mRNA therapy, and chemo/radiotherapy-mediated immunogenic cell death (ICD). Phototherapy, especially photodynamic therapy (PDT), has obvious advantages with highly localized treatment and minimal off-target effects by selectively activating photosensitizer-loaded tumor cells.
The nature of cell death determines CIC initiation. Apoptosis is an immune-silent cell death form with self-degradation and minimal release of cellular contents. In contrast, the lytic characteristics of programmed necrosis optimize antigen and adjuvant presentation, thus promoting CIC and generating strong tumor-specific T cell immunity. Therefore, it is urgent to utilize programmed necrotic cell death and amplify CIC through an effective cascade to achieve in situ cancer vaccination.
Optimize your cancer immunotherapy research with our high-purity CT26-OVA cells. Order your CT26-OVA cell now>>
2. Nano-photosensitizer Binding to Early Endosomal Membrane Induces Pyroptosis
Nano-bio interactions are critical for the intracellular fate and therapeutic effect of nanomedicines. Based on ultra-pH-sensitive nanotechnology, a series of amphiphilic copolymers mPEG5k-b-P(EPAx-r-DPA100–x) were synthesized, with x representing the repeat units of EPA monomers in the total number of EPA and DPA monomers. The pKa values of the synthesized copolymers ranged from 6.1 to 6.8, exactly matching the pH range of cancer cell early endosomes (EE). A series of PPa-conjugated copolymers were then prepared to explore the effect of chemical structure on nano-bio interactions and subsequent pyroptosis-inducing activity of EE-targeted nano-photosensitizers.
At pH 6.0, the membrane-binding capacity (R=0.97) and membrane rupture-induced LDH release capacity (R=0.83) of ionized nano-photosensitizers were positively correlated with EPA monomer units. The introduction of EPA monomers significantly enhanced the hemolytic effect of oxidative stress mediated by protonated nano-photosensitizers. Nano-probes with different pKa values exhibited adjustable subcellular distribution and biological functions. To exclude the influence of pKa, PPa-conjugated PEG5k-b-PDPA100 copolymer (PDPA) with a pKa of 6.5 and sharp pH response was synthesized for parallel comparison with PPa-conjugated mPEG5k-b-P(EPA50-r-DPA50) (PEPA). With similar subcellular transport behavior, PEPA had a 5.4-fold higher membrane-binding capacity and a 4.9-fold higher oxidative stress-induced hemolytic effect than PDPA at pH 6.0.
Negatively charged artificial lipid membranes (ALMs) were prepared to simulate the affinity between nano-photosensitizers and endolysosomal membranes. ITC studies showed that PEPA exhibited strong binding affinity to negatively charged ALMs at pH 6.0, but not at pH 7.4. The membrane-binding effect of nano-photosensitizers was also visualized by super-resolution microscopy. Ring-shaped fluorescence signals indicated that PEPA had higher binding affinity to endocytic organelle membranes than PDPA. When PEPA matured into early endosomes (PEPAEE), its superior endosomal membrane affinity significantly improved cellular photocytotoxicity. The half-maximal inhibitory concentration of PDPAEE was 3.5 times that of PEPAEE.
ROS stress in early endosomes activates the PLC-γ1/Ca2+/caspase-3 signaling axis, leading to the cleavage of gasdermin-E (GSDME) by caspase-3 and the release of its N-terminal fragment, which forms pores on the cell membrane to induce pyroptosis. Results including enhanced lipid peroxidation, PLC-γ1 phosphorylation, cytoplasmic calcium release, cellular caspase-3 activation, GSDME-N release, and pyroptotic phenotype with obvious cell swelling, giant vesicle formation and rapid increase in cell permeability confirmed that PEPAEE has efficient and specific pyroptosis-inducing ability.
PEPA was selected as the preferred candidate of EE-targeted nano-photosensitizer to study the role of endosome maturation-regulated pyroptosis in murine cancer cells. PEPA induced precise oxidative stress in early endosomes and lysosomes under 660 nm irradiation at 0.5 h and 2 h after cellular endocytosis, respectively. PEPAEE stress induced effective cell killing and presented a typical pyroptotic morphology, while PEPALy stress induced insufficient apoptosis in CT26 colorectal cancer cells with an IC50 value 4.9 times higher. GSDME knockout studies confirmed that PEPAEE stress induced strong GSDME-mediated pyroptosis. These results indicated that PEPA achieved switchable regulation of pyroptosis and apoptosis in CT26 cells through endosome maturation.
The PEPA-mediated regulable pyroptosis was further extended to a series of murine cancer cell lines with different GSDME expression levels. Interestingly, all eight GSDME-expressing cancer cell lines showed a 2.6 to 16.7-fold difference in regulable photocytotoxicity between PEPAEE and PEPALy, with a 3.8 to 5.0-fold difference in IC50 values. In addition, strong GSDME-mediated pyroptosis was observed in all cell lines after PEPAEE stress treatment.
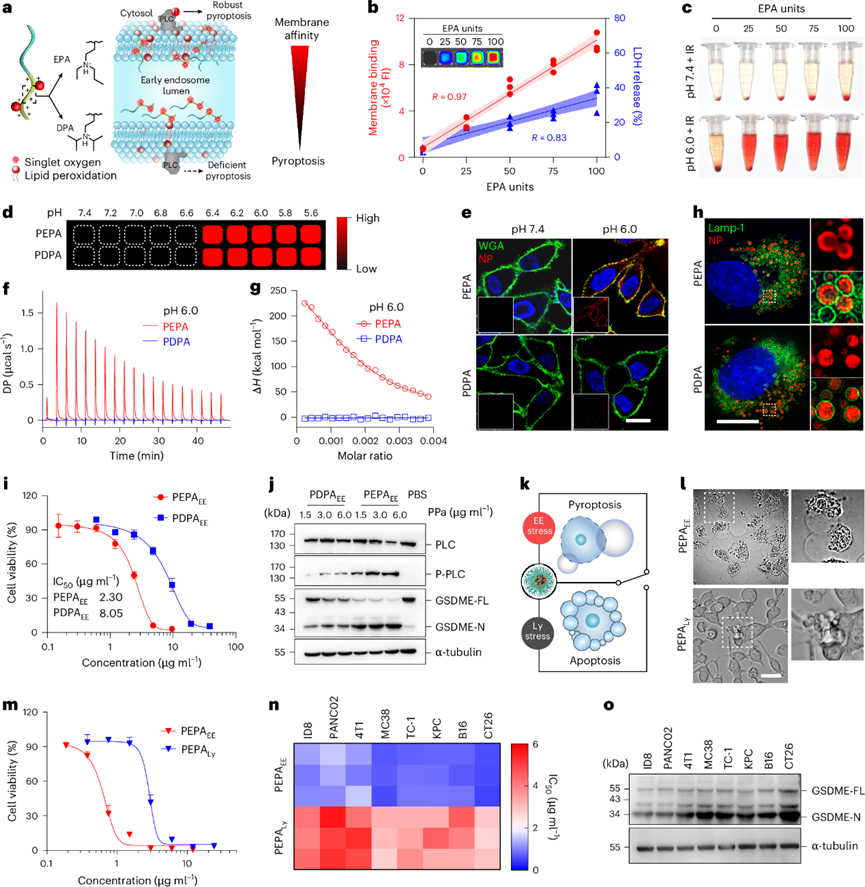
Fig. 1 Membrane affinity of nanoparticles regulates endoplasmic reticulum stress-induced pyroptosis
Our CT26-OVA cells are stably expressing ovalbumin. Contact us now to get CT26-OVA cell models and experimental protocols for your research!
3. EE Stress-induced Pyroptosis Enhances Tumor Immunogenicity
Stressed and dying cancer cells release a variety of bioactive molecules, including antigens and DAMPs. CT26 cells under PEPAEE stress had a 4.05-fold increase in protein release and a more diverse range of released proteins compared with cells under PEPALy stress. In particular, GSDME knockout effectively blocked PEPAEE-mediated release of cellular contents. Proteomic studies showed that PEPAEE induced the release of more protein copies. Gene ontology enrichment analysis revealed that these proteins were enriched in stress response and immune response related categories.
For altered proteins, significance analysis showed that 6 and 1136 proteins were decreased and increased in the supernatant of PEPAEE-stressed cells compared with that of PEPALy-stressed cells, respectively. Cluster analysis and KEGG enrichment analysis further found that PEPAEE significantly promoted the release of immunogenicity-related proteins and the activation of immune response-related signaling pathways, including immune cell recruitment, antigen presentation and innate immunity, compared with PBS and PEPALy. The enhanced immunogenicity of PEPAEE was also reflected in the significant increase of "find me" and "eat me" signals. In contrast, PEPALy stress and GSDME deficiency only slightly increased the immunogenicity of CT26 cells.
To explore the immune responses induced by different stressed cancer cells, murine bone marrow-derived dendritic cells were co-cultured with dying CT26-OVA cells. PEPALy-treated cells only induced weak DC activation, characterized by minimal upregulation of co-stimulatory markers (CD80 and CD86), minimal OVA (SIINFEKL) presentation and minimal cytokine (IL-12 and TNF-α) secretion. On the contrary, PEPAEE-stressed pyroptotic cells induced significant BMDC activation with a 10-fold higher cytokine secretion.
The organelle stress-mediated regulable immunogenicity and immune response were further evaluated in CT26-OVA tumor mouse models. Ultra-pH-sensitive nano-probes rapidly accumulated in tumor sites and were internalized by cells within 3 h after injection, with the peak colocalization with EEs at approximately 3 h. Western blot and immunofluorescence staining studies showed that PEPAEE stress successfully induced significant pyroptosis and ICD effects in in vivo tumor tissues. Therefore, the proportion of CD80+CD86+ DC cells presenting OVA antigen in the lymph nodes of mice treated with PEPAEE was higher than that of mice treated with PEPALy or CT26-OVA-GSDME−/− tumor mice.
DC maturation can trigger cytotoxic T cell-mediated adaptive immunity. In vivo specific cell killing studies showed that 91.2% of OVA-positive splenocytes were specifically eliminated in PEPAEE-treated mice, while this proportion decreased to 26.0% and 29.4% in PEPALy-stressed mice and CT26-OVA-GSDME−/− tumor mice, respectively. The excellent specific killing ability of PEPAEE in vivo further enhanced its tumor prevention ability when mice were rechallenged with CT26 cells, prolonging the median recurrence-free survival time by 1.6 times (25 days) and inhibiting tumor growth. These results indicated that the immunogenicity of cancer cells can be regulated by inducing different endocytic organelle stresses, and PEPAEE stress-mediated pyroptosis can effectively enhance ICD and cancer immune responses in vitro and in vivo.
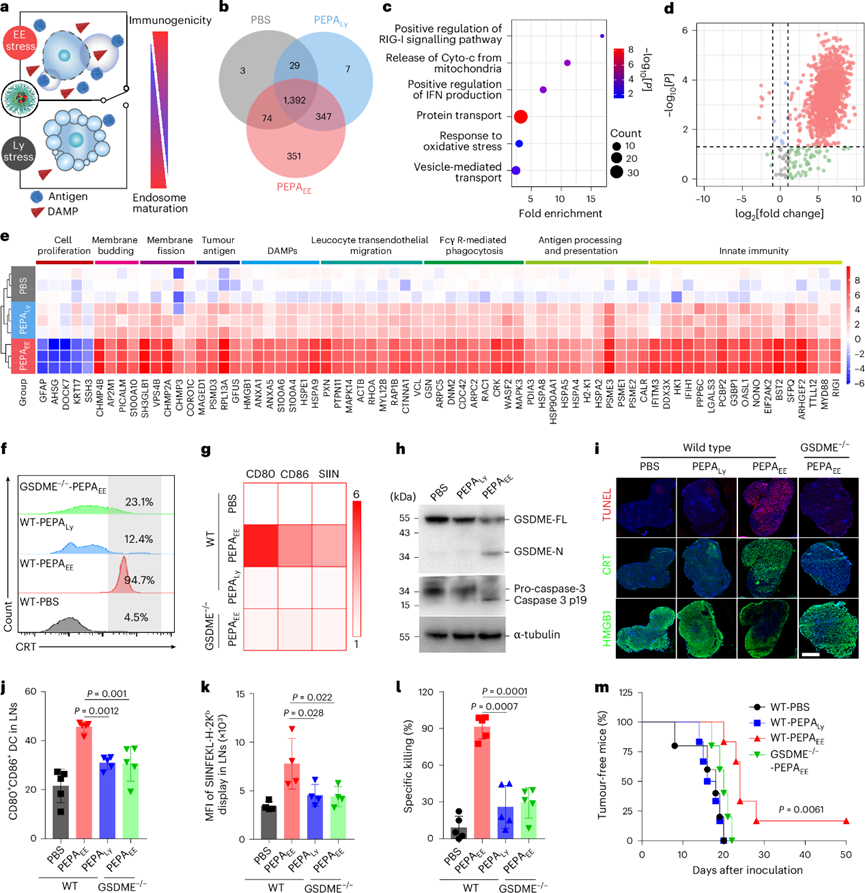
Fig. 2 Early endosome (EE) stress-induced pyroptosis modulates cancer cell immunogenicity
4. SPEN Design for Safe and Effective Immune Priming
Candidate vaccines need to have both antigenicity and adjuvanticity. Although PEPAEE-induced pyroptotic cells have abundant antigenicity, the adjuvanticity of released DAMP molecules is insufficient for effective cancer vaccination. Therefore, a light-controlled and ROS-sensitive SPEN nanoadjuvant was constructed by conjugating a TLR7/8 agonist IMDQ to the hydrophobic block of PEPA via a thioketone bond. These polymer-drug conjugates can self-assemble into nanoadjuvants.
More than 80% of IMDQ in SPEN was released under 660 nm laser irradiation, while no drug release was observed in SPIN. The light-controlled immune activation ability of SPEN was studied using TLR reporter cell line (Raw-Blue) and murine BMDC. SPEN irradiated with 660 nm laser exhibited potent TLR activation activity equivalent to free IMDQ (half-maximal effective concentration EC50=85 nmol). In vivo studies showed that PEPA and SPEN exhibited similar pharmacokinetic characteristics and long circulation properties after intravenous injection, with β-phase half-lives of 14.9 h and 12.3 h, respectively.
In vivo distribution and tumor imaging studies showed that SPEN was rapidly internalized by cells and activated in tumors within 3 h after injection. The intratumoral SPEN signal at 3 h after injection was 53.2% of that at 24 h. Ex vivo imaging showed that SPEN specifically accumulated and was activated in tumor tissues, with a signal intensity 1.8 times and 5.7 times that of liver and spleen, respectively. Flow cytometry analysis further showed that 14.9% of cells in tumors internalized SPEN, with tumor cells accounting for 38.8% of this population. In contrast, the uptake of SPEN in the spleen was negligible.
The low activation of IMDQ and ROS-gated release jointly inhibited the innate immune activation function of SPEN in the liver and spleen. Importantly, SPEN effectively eliminated the adverse systemic and immune toxicity caused by intravenous injection of free IMDQ, characterized by minimal weight loss, splenomegaly, cytokine storm and cytotoxicity to major organs. Therefore, the oxidative stress-gated design of SPEN enables systemic delivery of immune adjuvants and can safely and specifically prime tumor immunity.
5. SPEN-mediated EE Stress Forms Pyroptosomes as Cancer Vaccines
Cancer cell membranes have been widely used as carriers and antigen sources for cancer vaccines. The vaccine efficacy of SPENEE-induced pyroptotic cells mediated by SPEN was then studied. Micron-sized pyroptosomes were obtained by purification through differential centrifugation. CT26-OVA cells transfected with Rab5a-GFP were stressed with SPEN encapsulated with 10 nm gold nanoparticles (AuNP). EEs, internalized SPEN and IMDQ were enriched in pyroptosomes. Western blot analysis of cell membranes further showed that the contents of cleaved gasdermin D, calreticulin and OVA in pyroptosomes were 5.1 times, 2.5 times and 2.9 times higher than those in untreated cell membranes, respectively.
The in vitro and in vivo vaccination effects of pyroptosomes were then explored. Benefiting from its superior TLR stimulation activity and higher adjuvanticity, SPENEE significantly enhanced the maturation and antigen presentation ability of BMDC by EE stress-induced pyroptosomes. After injection into the footpad, pyroptosomes exhibited rapid, efficient and long-lasting lymph node drainage capacity. The fluorescence signal in the popliteal lymph nodes of mice injected with DiR-labeled pyroptosomes was 6.3 times higher than that of mice injected with lysosome-stressed cells. Flow cytometry studies further showed that 69.0% of DiR-positive cells were dendritic cells and macrophages, indicating that most pyroptosomes were internalized by APCs.
Immunization with pyroptosomes further led to lymph node enlargement with a 4.0-fold increase in weight. The proportion of mature dendritic cells in the lymph nodes of mice immunized with SPENEE or PEPAEE-induced pyroptosomes was 4.1 times and 3.1 times higher than that of unimmunized mice, respectively, and the OVA presentation level was also significantly increased. The immune response in lymph nodes was further verified by immune cell proliferation, infiltration of antigen-presenting cells in the paracortical T cell area and migration of CD4 T cells to the germinal center. These results indicated that pyroptosomes can effectively deliver tumor antigens and adjuvants to lymph nodes and function as highly immunogenic cancer vaccines.
The cancer prevention efficacy of pyroptosome-based vaccines was then studied. Immunization with SPENEE-induced pyroptosomes derived from CT26 cells successfully inhibited CT26 tumor growth in a dose-dependent manner. When BALB/c mice were treated with pyroptosomes derived from 2×105 pyroptotic cells, the tumor volume was reduced to 1.6%, and the tumor incidence was 60% on day 22 after tumor cell inoculation. However, the cancer vaccine efficacy of pyroptosomes was lost in immunodeficient nude mice.
Autologous tumors have been used to produce cancer vaccines for personalized cancer immunotherapy to prevent postoperative recurrence and metastasis. Therefore, spontaneous breast tumors were resected from MMTV-PyMT/FVB mice, and the SPENEE-based pyroptosome vaccine strategy was applied. After immunization with autologous tumor-derived pyroptosomes, the occurrence of mammary hyperplasia and tumors in transgenic mice was significantly inhibited. Mice not immunized with pyroptosome vaccine developed approximately 8.5 tumors per mouse with a total weight of 1.6 g. In contrast, mice immunized with pyroptosome vaccine developed an average of 2.8 tumors per mouse (32.9%) with a relatively low total weight (0.17 g, 10.6%).
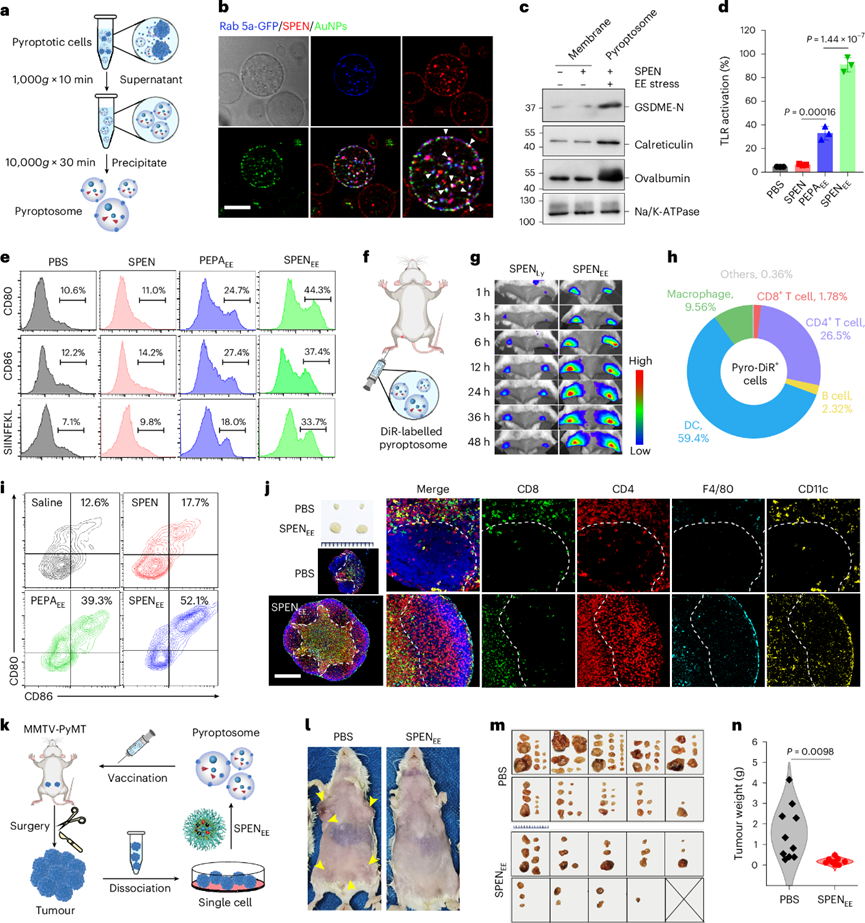
Fig. 3 SPEN-mediated SPENEE-derived pyroptosomes function as cancer vaccines
6. Pyroptosomes Enhance the Cancer-Immunity Cycle
After confirming the immunization function of pyroptosomes, the potential mechanism of SPEN-mediated vaccination was then clarified. In vivo specific killing studies showed that CT26-OVA tumor mice treated with PEPAEE or SPINEE mediated by SPIN exhibited OVA-specific cell killing rates of 81.1% and 84.6%, respectively. With the immune priming effect of IMDQ adjuvant, SPENEE stress achieved a 99.7% OVA-specific killing effect.
Compared with negligible cytotoxic CD8 T cells in unimmunized mice, the number of cytotoxic CD8 T cells expressing granzyme B and IFN-γ was significantly increased in all mice treated with EE stress. In mice immunized with SPENEE stress, the proportions of granzyme B+/CD8+ T cells and IFN-γ+/CD8+ T cells were increased to 16.7 times and 6.0 times those of unimmunized mice, respectively. These results indicated that SPENEE stress-mediated pyroptosis initiated adaptive T cell immune responses in vivo.
To clarify the intratumoral immune mechanism, the tumor immune microenvironment was further visualized and analyzed by multicolor fluorescent immunohistochemistry. SPENEE stress successfully remodeled the tumor immune microenvironment, and the populations of tumor-suppressive immune cells such as CD8+ T cells, NK cells and M1-type macrophages were increased to 20.3 times, 14.8 times and 35.2 times, respectively. Although a slight increase in immunosuppressive Foxp3+ Tregs was observed in immunized mice, the ratios of CD8+ T cells to Foxp3+ T cells and M1-type to M2-type macrophages were significantly expanded, further indicating that SPENEE-mediated in vivo vaccination can reverse the immunosuppressive state in the tumor microenvironment.
In addition, the TLR7/8 agonist incorporated in SPENEE effectively offset the secondary immunosuppression caused by phototherapy, characterized by negligible upregulation of immune checkpoint molecules on tumor cells (PD-L1 and CD47) and T cells (PD-1), and no detectable autoimmune response was induced even in IL-10-deficient mice with spontaneous colitis. Macrophages and NK cells play key roles in innate immunity. SPENEE-mediated in situ vaccination achieved 100% cancer prevention effect, while depletion of CD8+ T cells, NK cells and macrophages led to rapid tumor recurrence in immunized mice.
The effect of pyroptosomes on macrophages and NK cells was then studied in vitro. Macrophages are one of the main antigen-presenting cells. Previous studies have confirmed that IMDQ-based nanoadjuvants can selectively reprogram M2-type tumor-associated macrophages with antigen-destructive effects into M1 phenotype with improved antigen presentation ability. SPENEE-mediated pyroptosomes and their released IMDQ significantly improved the presentation of MHC-I and OVA antigens on the membrane of M2-type macrophages. In addition, immature NK cells derived from the spleen were successfully activated into effector NK cells with cytotoxic function by pyroptosomes, characterized by increased membrane expression of NKp46 and CD107a, leading to a 77.7% killing rate of NK cells on CT26 cells. These results indicated that SPEN-mediated in situ vaccination activates innate and adaptive cancer immunity, thus closing the CIC.
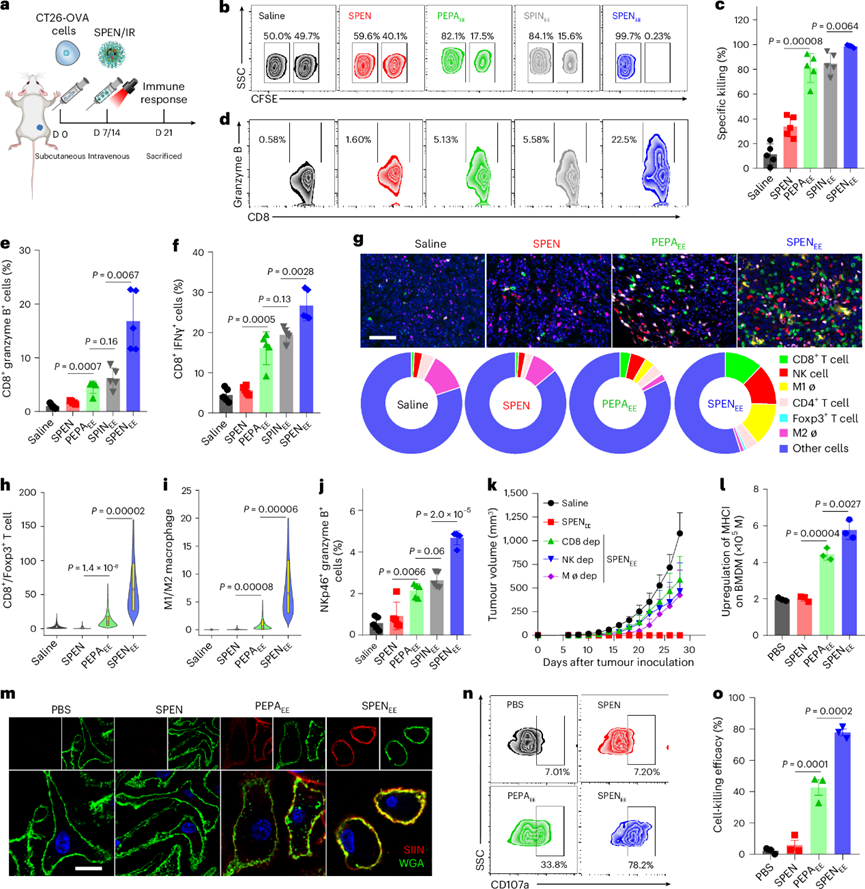
Fig. 4 SPENEE reshapes the anti-tumor immune microenvironment in situ
7. SPENEE-driven In Situ Vaccination Facilitates Cancer Therapy
Given the satisfactory immunization ability of SPENEE, it was applied to in vivo cancer immunotherapy. First, the abscopal effect was evaluated in the CT26 cancer model. The results showed that SPENEE stress significantly eradicated irradiated and abscopal tumors, with tumor-free rates of 88.9% (8/9) and 77.8% (7/9), respectively. Without the effect of IMDQ adjuvant, the efficacy of PEPAEE and SPINEE was affected, especially the abscopal effect, with tumor-free rates reduced to 11.1% (1/9) and 0% (0/9), respectively. The survival time of tumor mice treated with SPENEE was also significantly prolonged.
The therapeutic effect was then evaluated in the B16 melanoma model. Although PEPAEE stress eliminated most tumor burdens, 87.5% of mice relapsed rapidly during the drug withdrawal period. In contrast, 75% of mice with large tumors were completely cured after SPENEE treatment, with no animal death and lymph node metastasis during the two-month treatment-free period.
To further verify the preventive and therapeutic activities of SPENEE-mediated in situ vaccine, SPENEE stress immunization was performed on MC38 colorectal tumor mice. Immunized mice exhibited significant anti-tumor effects with a 100% (10/10) tumor regression rate. Importantly, all tumor-free mice in the SPENEE group successfully resisted multiple MC38 tumor rechallenges (day 30, day 60 and day 90) during the 5-month treatment-free period. Long-term cancer prevention for more than 150 days indicated that SPENEE-mediated in situ vaccine induced strong immune memory. This was further confirmed by a 5.1-fold and 3.4-fold increase in central memory CD8+ T cells and CD4+ T cells, respectively, and a significant increase in effector memory T cells compared with the PBS group.
Benefiting from its excellent vaccination effect, SPENEE stress also significantly prevented lung metastasis in the highly invasive 4T1 tumor model, characterized by no detectable luminescent signal in lung tissues. Considering the poor efficacy of immune checkpoint blockade therapy for "cold" tumors with immune-silent phenotype, SPENEE-mediated in situ vaccine strategy was combined with αPD-1 to enhance the therapeutic effect in the Panc02 pancreatic cancer model. αPD-1 monotherapy had negligible tumor suppressive effect on mice, while 50% of SPENEE-immunized mice showed no tumor progression. In particular, the combination therapy of SPENEE and αPD-1 achieved 83.3% eradication of primary tumors, and significantly prolonged the survival time of mice and prevented cancer within 120 days.
To improve clinical relevance and influence, the immunotherapeutic effect of the combination strategy was further evaluated using tumor models with large abscopal tumors or multiple tumor nodules. In CT26 tumor mice, SPENEE had a similar inhibitory effect on PDT tumors, and combination with αPD-1 effectively improved the eradication rate of large abscopal tumors with a tumor-free rate of 80%. In addition, the strong systemic anti-tumor immunity generated by SPENEE-mediated in situ vaccination combined with PD-1 blockade successfully regressed pre-established multiple peritoneal CT26-luc tumor metastases, with a significant reduction in metastatic tumor nodules and luminescent signals.
A lung metastasis model was also established by intravenous injection of 4T1-luc cells before treatment, characterized by a rapid increase in luminescent signals in the lungs. In contrast, the luminescent signals and metastatic tumor nodules in the lungs of mice treated with SPENEE combined with αPD-1 were negligible. Finally, the combination strategy was applied to 8-week-old MMTV-PyMT mice with multiple spontaneous hyperplasias and tumors. The tumor in the left cervical mammary gland was selected as the PDT tumor and irradiated with 660 nm laser 3 h after SPEN injection. Mice treated with normal saline developed an average of 8 tumors per mouse with a total tumor weight of 3.5 g. After combination therapy, the tumor burden was significantly reduced. Mice developed an average of 3.3 tumors per mouse (58.8% reduction) with a total tumor weight of 0.33 g (90.6% reduction). In conclusion, the EE-regulated immunogenic pyroptosis combined with adjuvant activation platform provides a promising in situ vaccine strategy for precise cancer immunotherapy.
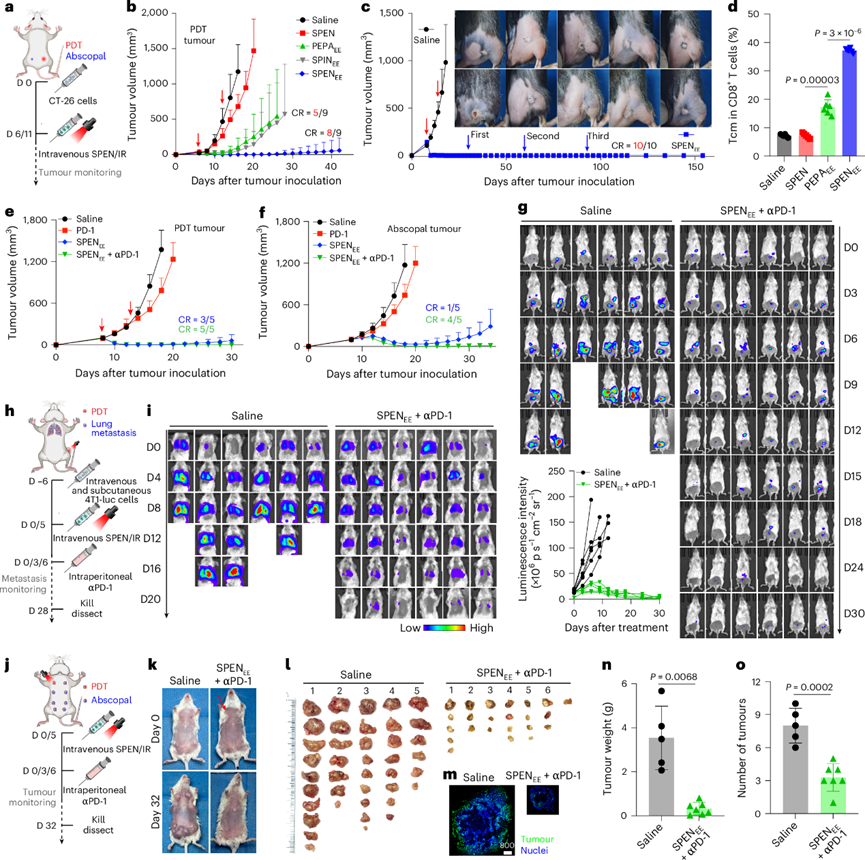
Fig. 5 SPENEE enables potent tumor immunotherapy with durable vaccination effects across multiple cancer types
8. Conclusion
Weak immunogenicity and poor accessibility of patient-specific tumor antigens are the main obstacles to the clinical efficacy of cancer vaccines. In situ cancer vaccines utilizing autologous tumor antigens at tumor sites have been proposed as personalized cancer immunotherapies after surgery. To optimize the immunogenicity of in situ tumor vaccines, a variety of strategies including chemotherapy, radiotherapy, phototherapy and oncolytic virus therapy have been used as cell stress inducers to trigger ICD.
However, the immunological correlation between cancer immunity and organelle stress-induced ICD effects, as well as their potential immunogenic mechanisms, remain unclear. Benefiting from spatiotemporally adjustable pyroptosis nanotechnology, the research team successfully revealed the organelle stress-dependent amplification effect of cancer cell immunogenicity in vitro and in vivo. A SPEN that can enhance cellular immunogenicity was further developed, which can function as an in situ cancer vaccine. This study not only confirmed the encouraging immunogenic function of EE stress-induced pyroptosis and revealed the potential immune priming mechanism of pyroptosis-derived vesicles, but also opened up new opportunities for the design of pyroptosis-inducing nanomaterials for personalized cancer immunotherapy.
References
Chen, B., Wan, F., Xia, H., et al. In situ-generated vaccine-like pyroptosome for personalized cancer immunotherapy. Nature Materials. 2026 Mar 3; https://doi.org/10.1038/s41563-026-02506-9
