MDA-MB-231-Luc: CDK16-Mediated PRC1 Phosphorylation Drives Metastasis in Triple-Negative Breast Cancer
Introduction
Triple-negative breast cancer (TNBC) remains the most clinically challenging breast cancer subtype, defined by the absence of estrogen receptor (ER), progesterone receptor (PR), and HER2 amplification. Accounting for 15–20% of all breast cancers, TNBC is disproportionately aggressive, with high rates of distant metastasis and poor overall survival. Cyclin-dependent kinase 16 (CDK16), an atypical PCTAIRE kinase activated by Cyclin Y (CCNY) family members, has recently emerged as a critical driver of TNBC malignancy. Using MDA-MB-231-Luc cells engineered to stably express firefly luciferase, researchers demonstrated that CDK16 overexpression dramatically accelerates in vivo tumor growth and pulmonary metastasis, detectable by non-invasive bioluminescence imaging (BLI). These findings establish CDK16 as both a mechanistic driver and a promising therapeutic target in TNBC.
Looking for a high-performance TNBC model for metastasis and drug efficacy studies? Our MDA-MB-231-Luc cells deliver stable, sensitive bioluminescence signals for real-time in vivo tumor tracking.
Order now: MDA-MB-231 Cell Line / MDA-MB-231 Luciferase Cell Line / MDA-MB-231-Luc-GFP Cell Line
CDK16 Is Overexpressed in TNBC and Correlates with Poor Prognosis
Analysis of clinical datasets revealed that CDK16 is significantly upregulated in basal-like and TNBC tumors compared with other breast cancer subtypes and normal tissue. High CDK16 expression correlated with shorter overall survival, higher histological grade, and increased metastatic recurrence risk. Importantly, CDK16 transcript levels were elevated in MDA-MB-231 cells relative to luminal cell lines (MCF-7, T47D), underscoring the TNBC-specific relevance of this kinase.
CDK16 Knockdown Suppresses TNBC Tumor Growth In Vitro and In Vivo
Stable shRNA-mediated knockdown of CDK16 in MDA-MB-231-Luc and SUM159 cells significantly reduced colony formation, cell viability (CCK-8 assay), and 3D spheroid growth. In xenograft mouse models, CDK16 knockdown led to a marked reduction in tumor volume and weight compared with control cells. Crucially, reintroduction of wild-type CDK16 — but not a kinase-dead mutant (D304A) — rescued proliferation, demonstrating that kinase activity is indispensable for CDK16-driven tumor growth.
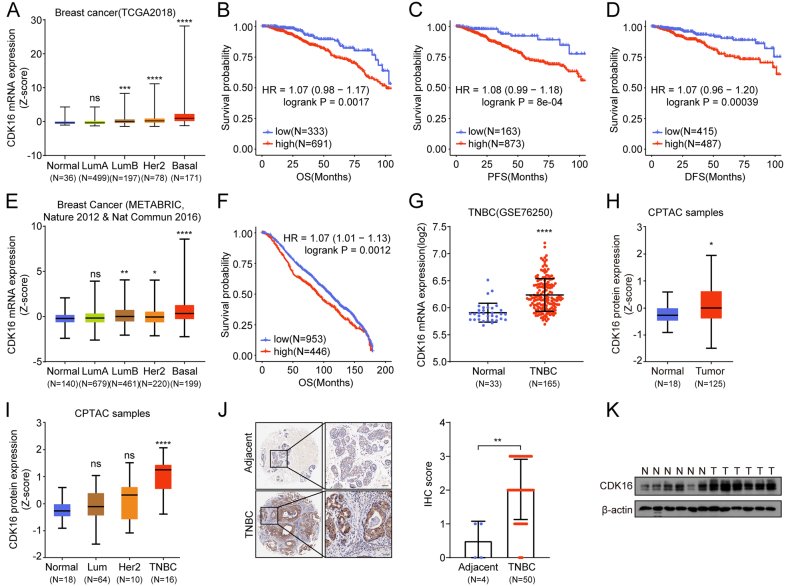
Figure 1. CDK16 knockdown inhibits TNBC tumor growth in vitro and in vivo. (A) Colony formation assay following CDK16 shRNA knockdown. (B) Tumor growth curves in CDX xenograft models. (C) Tumor weight comparison. (D) Kinase-dead mutant rescue experiment.
Validation in Patient-Derived Organoid and Xenograft Models
To strengthen clinical translatability, CDK16 knockdown was validated in patient-derived organoid (PDO) and patient-derived xenograft (PDX) models from TNBC patients. CDK16 silencing significantly reduced organoid formation efficiency and Ki-67 proliferative index in PDO models. In PDX experiments, CDK16-depleted tumors grew substantially slower, with histological analysis confirming decreased mitotic figures. These results demonstrate CDK16 dependency extends beyond established cell lines to primary patient tumor biology.
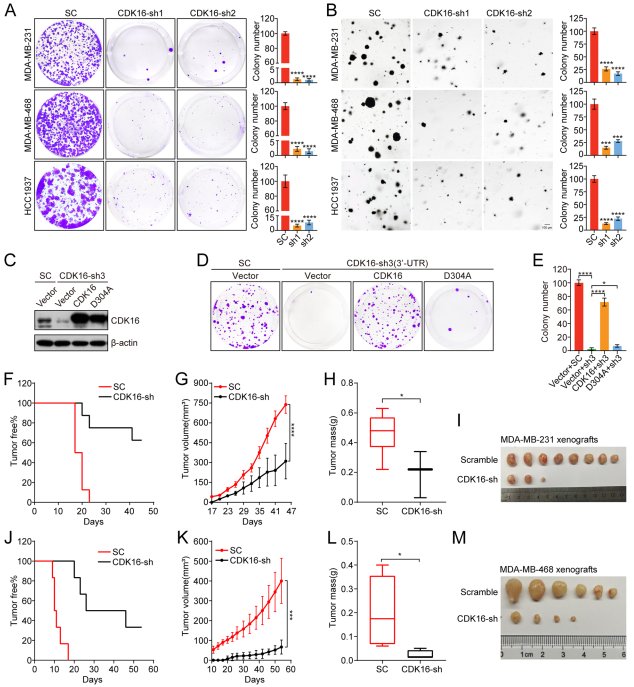
Figure 2. CDK16 knockdown suppresses tumor growth in PDO and PDX models. (A) Organoid formation efficiency. (B) Representative PDX tumor images. (C) Tumor growth curves. (D) Ki-67 immunohistochemistry.
CDK16 Promotes Invasion and Pulmonary Metastasis via Bioluminescence-Tracked Models
Using MDA-MB-231-Luc cells with stable CDK16 overexpression or knockdown, researchers quantitatively monitored metastatic progression through weekly bioluminescence imaging (BLI) in tail vein injection models. CDK16 overexpression significantly increased BLI signal intensity in lungs and distant organs, corresponding to elevated pulmonary metastatic nodule counts at necropsy. Transwell invasion and wound-healing scratch assays confirmed that CDK16 knockdown suppressed in vitro migratory and invasive capacity of MDA-MB-231-Luc cells, establishing a direct link between CDK16 activity, cytoskeletal dynamics, and metastatic dissemination.
Accelerate your TNBC metastasis research with our MDA-MB-231-Luc cells — validated for stable luciferase expression across passages and proven performance in pulmonary metastasis models. Request a quote now to get started.
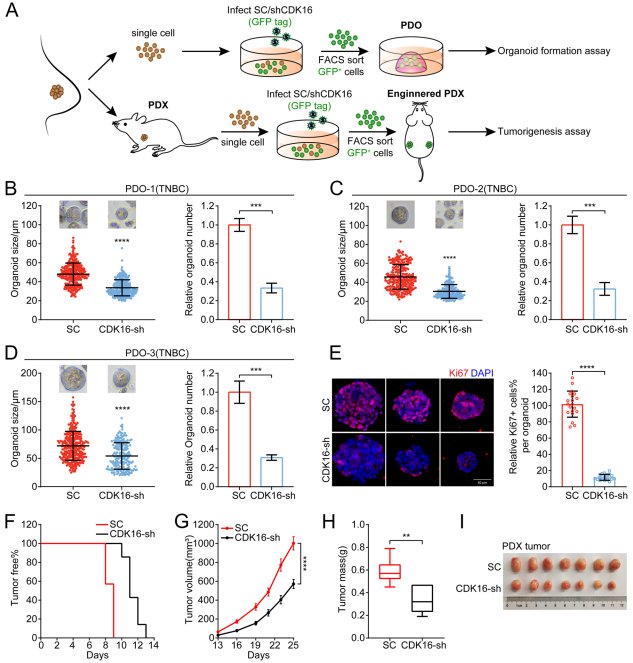
Figure 3. CDK16 promotes TNBC invasion and pulmonary metastasis. (A) Bioluminescence imaging of lung metastasis at weeks 2, 4, 6. (B) BLI signal quantification. (C) Lung metastatic nodule counts. (D) Transwell invasion assay results.
Mechanism: CDK16 Phosphorylates PRC1 at Thr481 to Dysregulate Spindle Assembly
Through mass spectrometry-based phosphoproteomic screening combined with co-immunoprecipitation, PRC1 (protein regulator of cytokinesis 1) was identified as a direct CDK16 substrate. CDK16 phosphorylates PRC1 at threonine 481 (T481), disrupting PRC1 nuclear-cytoplasmic shuttling and impairing central spindle assembly during mitosis. In MDA-MB-231 cells, CDK16 overexpression led to abnormal spindle morphology and increased mitotic errors, while CDK16 knockdown restored ordered spindle formation. Phospho-mimetic PRC1 (T481E) recapitulated CDK16 overexpression phenotypes, while the non-phosphorylatable T481A mutant abrogated CDK16-driven proliferation, formally placing PRC1 phosphorylation downstream of CDK16 in the oncogenic pathway.
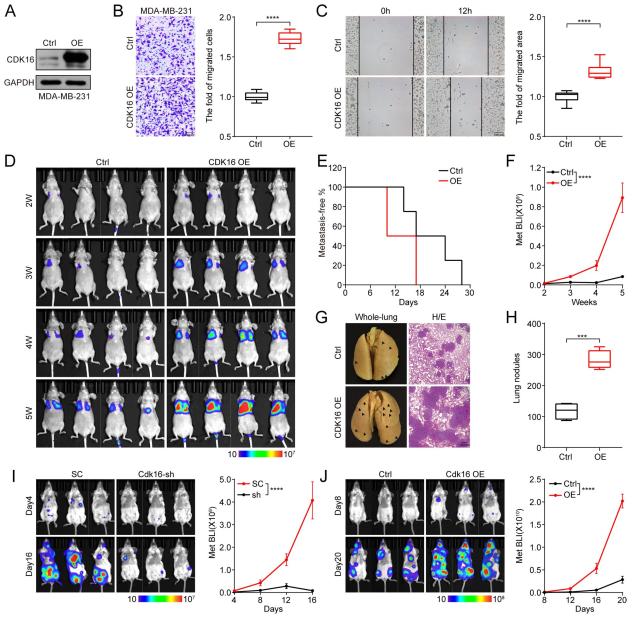
Figure 4. CDK16 phosphorylates PRC1 at T481 to dysregulate spindle assembly. (A) PRC1 identified as CDK16 substrate by mass spectrometry. (B) Immunofluorescence showing abnormal spindle morphology. (C) PRC1 nuclear-cytoplasmic localization changes. (D) Phospho-mutant rescue experiments.
Therapeutic Targeting: CDK16 Inhibitor Rebastinib Suppresses Tumor Growth and Metastasis
Rebastinib (Reb), identified as a CDK16 inhibitor through kinase profiling, effectively reduced PRC1 T481 phosphorylation in a dose-dependent manner. In MDA-MB-231-Luc xenograft models, Reb administration significantly inhibited tumor growth and reduced BLI-measured metastatic burden, outperforming CDK4/6 inhibitor abemaciclib (Abe). In PDO models, Reb suppressed organoid formation with IC50 values in the nanomolar range. Transcriptomic analysis confirmed that CDK16 inhibition broadly downregulated mitotic spindle assembly, G2/M checkpoint, and Rb-E2F pathway gene signatures, revealing multi-pathway anti-tumor mechanisms.
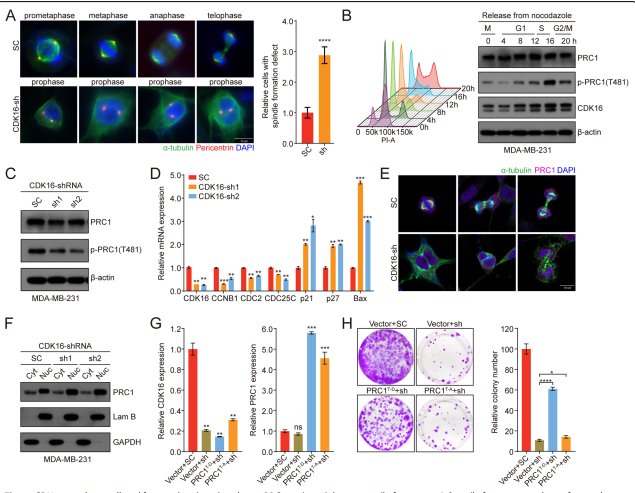
Figure 5. CDK16 inhibitor Rebastinib suppresses tumor growth and metastasis. (A) Tumor growth curves in xenograft models. (B) BLI-measured metastatic burden. (C) PDO IC50 determination. (D) Comparison with abemaciclib.
Conclusion
This study establishes CDK16 as a key oncogenic kinase in TNBC, acting through PRC1 T481 phosphorylation to disrupt spindle assembly and promote metastatic dissemination. The MDA-MB-231-Luc bioluminescent cell model proved instrumental in quantitatively capturing CDK16-driven metastatic progression in real time, providing robust pharmacodynamic endpoints for the CDK16 inhibitor rebastinib. These findings nominate the CDK16-PRC1 axis as a high-priority therapeutic target in TNBC and validate MDA-MB-231-Luc as the gold-standard model for preclinical evaluation of anti-metastatic strategies.
Ready to power your TNBC drug discovery pipeline? Our MDA-MB-231-Luc cells combine triple-negative biology with real-time bioluminescent metastasis tracking. Browse our catalog and find the perfect cell for your study.
Request a quote now: MDA-MB-231 Cell Line / MDA-MB-231 Luciferase Cell Line / MDA-MB-231-Luc-GFP Cell Line
References
Huang Z, et al. CDK16 phosphorylates and destabilizes PRC1 to promote tumor progression and metastasis in triple-negative breast cancer. J Exp Clin Cancer Res. 2022 Apr;41(1):141. doi: 10.1186/s13046-022-02362-w
